

Резистентность к ингибиторам тирозинкиназы EGFR:
проблема и пути ее решения

Рак легкого был и остается самой частой причиной смерти от злокачественных заболеваний в мире, при этом немелкоклеточный рак легкого (НМРЛ) регистрируется в 85% случаев [1]. Химиотерапия, составляющая основу лечения НМРЛ, не всегда демонстрирует достаточную эффективность в отношении данного заболевания [2]. Препараты нового класса, ингибиторы тирозинкиназы (ИТК) EGFR, целью которых являются рецепторы эпидермального фактора роста (EGFR), в частности, препарат гефитиниб, и в клинических испытаниях, и на практике показали многообещающие результаты [3]. Чувствительность к ним определяется наличием в опухолевых клетках соматических мутаций в гене EGFR, представляющих собой небольшие делеции аминокислот 747-750 или точечные мутации (наиболее часто в виде замены лейцина на аргинин в кодоне 858 (L858R)) [4]. Было показано, что применение ИТК у пациентов с подтвержденной мутацией в гене EGFR ассоциируется с увеличением частоты объективных ответов, а также способствует повышению продолжительности жизни без прогрессирования [5]. Тем не менее, успех терапии ИТК НМРЛ с мутацией в гене EGFR в подавляющем большинстве случаев носит временный характер: как правило, после года терапии заболевание начинает прогрессировать [6].
Понятие первичной и приобретенной резистентности
Устойчивость, или резистентность к воздействию химиотерапевтических лекарственных средств, является одним из основных ограничений для лечения онкологических заболеваний, в том числе и немелкоклеточного рака легких (НМРЛ). Опухоли могут демонстрировать как резистентность de novo, то есть первичную, так и вторичную, приобретенную устойчивость.
Первичная резистентность имеет место в случаях, когда некоторые присущие раковым клеткам первичные характеристики обусловливают ее устойчивость к данному классу лекарственных препаратов.
Так, около 25% пациентов с НМРЛ с мутациями EGFR в опухолевых клетках не отвечают на терапию ИТК (по сравнению с 90% среди всех больных НМРЛ) [6]. Известно, что некоторые мутации EGFR, расположенные в экзонах 18-21, ассоциируются с первичной резистентностью к ИТК. Например, небольшие инсерции или дупликации в экзоне 20 (такие как D770_N771, NPG, SVQ и другие), которые наблюдаются в 5% случаев НМРЛ, менее чувствительны к ИТК, чем опухоли с мутациями в экзоне 19 и L858R [7].
Первичная резистентность к ИТК может также быть результатом и более редких мутаций в EGFR, которые встречаются наряду с мутациями, ответственными за чувствительность к лекарственным препаратам.
Приобретенная резистентность возникает в случаях, когда первично чувствительные к лекарственному препарату опухолевые клетки становятся устойчивыми к терапии уже во время лечения [8].
Причины приобретенной резистентности к ИТК EGFR
Одной из наиболее распространенных причин приобретенной устойчивости к ИТК является вторичная мутация в экзоне 20, которая заключается в замещении метионина на треонин в положении 790 (Т790М) в домене киназы. Мутации Т790М в EGFR регистрируются более чем в 50% опухолей с приобретенной резистентностью к ИТК [9]. Наряду с Т790М приобретенная устойчивость может быть ассоциирована с другими вторичными мутациями, в том числе L747S (экзон 19) [10], D761Y (экзон 19) [11] и Т854А (экзон 21 в петле активации) [12].
Примерно в 20% случаев вторичной устойчивости к терапии ИТК ее причиной является амплификация онкогена МЕТ, при этом не имеет значения, существует ли мутация Т790М, или нет [13]. Рецептор тирозинкиназы МЕТ мутирует или избыточно экспрессируется при многих опухолях, в том числе НМРЛ. МЕТ образуется главным образом на клетках эпителиального происхождения и активируется его лигандом, фактором роста гепатоцитов (HGF), отвечая за запуск ряда внутриклеточных сигнальных каскадов [14]. МЕТ и HGF необходимы для эмбрионального развития, так как они участвуют в обнаружении дефектов в эпителиально-мезенхимальном переходе во время органогенеза [15]. Кроме того, они играют важную роль в клеточной пролиферации, ангиогенезе, миграции и инвазии клеток, а также морфогенной дифференциации и тканевой организации.
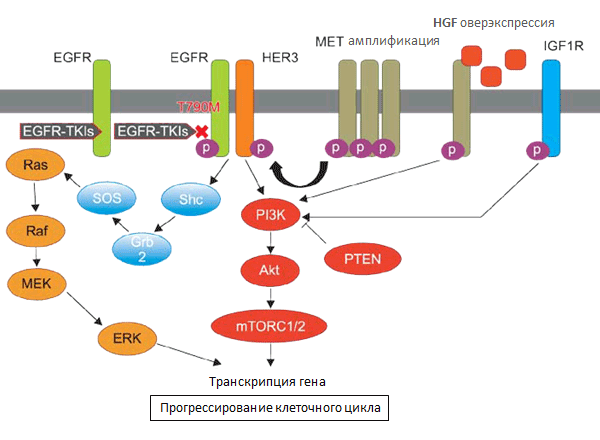
Функция МЕТ в опухолях человека может быть повышена за счет амплификации этого рецептора, что приводит к серьезным изменениям, в том числе, пролиферации опухолевых клеток, снижению апоптоза, ангиогенезу, изменению функции цитоскелета и метастазированию, а также развитию устойчивости к ИТК.
Опухоли примерно 40% пациентов с приобретенной резистентностью к ИТК не имеют вторичной мутации Т790М или амплификации МЕТ. Существуют данные, подтверждающие роль тирозиновой протеинкиназы EGFR/ErbB, которая кодируется геном ERBB2, HER2 в развитии резистентности к ИТК. Так, в работе Ken Takezawa [16] продемонстрировано, что в 12% случаев приобретенной устойчивости была зафиксирована амплификация HER2 (по сравнению с 1% у пациентов с нелеченной аденокарциномой легких).
Известны случаи, когда у больных с изначально диагностированной аденокарциномой с мутацией EGFR и развившейся приобретенной резистентностью к ИТК во время рецидива был выявлен мелкоклеточный рак легкого (МРЛ) [17]. До сих пор остается неизвестным, вызвана ли трансформация НМРЛ в МРЛ наличием в первичной опухоли компонента мелкоклеточного рака, или процесс ассоциирован с трансдифференциацией аденокарциномы.
In vitro была показана связь между эпителиально-мезенхемальным переходом (ЕМП) и устойчивостью к ИТК EGFR [18]. Более того, данные исследований свидетельствуют, что ЕМП можно выявить и в опухоли пациента [19]. С приобретенной резистентностью может быть ассоциировано и усиление сигнализации от рецептора инсулин-подобного фактора роста (IGF1R), хотя эта связь была продемонстрирована только в модели in vitro с использованием линии клеток, экспрессирующей высокий уровни «дикого» типа EGFR [20]. Следует подчеркнуть, что в механизме развития приобретенной резистентности к ИТК EGFR остается немало белых пятен, и активные исследовательские работы в этой области продолжаются. Одним из наиболее изученных на сегодняшний день механизмов формирования резистентности является самый распространенный – вторичная мутация Т790М.
Сегодня известны по крайней мере два молекулярных механизма, объясняющие, каким образом мутация Т790М придает опухоли лекарственную устойчивость. Во-первых, замена метионина на треонин в положении 790 приводит к изменению связывания препарата в кармане АТФ EGFR. Во-вторых, мутация Т790М значительно увеличивает афинность АТФ к EGFR-L858R, практически восстанавливая сродство АТФ до уровня «дикого» типа EGFR. Это восстановление закрывает терапевтическое окно, возникающее вследствие сниженной афинности АТФ к онкогенным мутациям, которые обычно более легко ингибируются по сравнению с диким EGFR [21]. Развитие приобретенной устойчивости вследствие мутации Т790М может происходить двумя путями. В первом случае замещение метионина на треонин в Т790М в опухолевых клетках отсутствует изначально и развивается de novo в одной или более клональной популяции на фоне лечения ИТК EGFR. Во второй модели замещение существует в cis вследствие первичной активирующей мутации у небольшой популяции и «проявляется» в присутствии ИТК EGFR.
Биохимические исследования свойств Т790М показали, что у пациентов с приобретенной резистентностью на фоне вторичной мутации усиливается активность киназы к первично активированным аллелям EGFR, что, возможно, способствует повышению онкогенности [22]. Однако, несмотря на это, больные с мутацией Т790М могут демонстрировать достаточно медленные темпы прогрессирования заболевания [23]. Более того, даже после прекращения терапии ИТК в некоторых случаях регистрируется положительная динамика в развитии заболевания [24]. Это указывает на то, что часть опухолевых клеток все-таки остается чувствительной к ингибированию EGFR. В подтверждение этой теории сообщается о множественных повторных ответах на терапию ИТК после короткого перерыва в таргетной терапии [24]. Добавим, что точный механизм подобного явления до сих пор остается невыясненным.
Возможности лечения НМРЛ у пациентов с приобретенной резистентностью
Учитывая актуальность проблемы резистентности к ИТК, альтернативная длительная стратегия терапии НМРЛ с мутацией EGFR должна учитывать необходимость предотвращения или хотя бы увеличения времени до развития устойчивости. Один из подходов к рационализации терапии заключается в изучении влияния различных стратегий дозирования с использованием уже существующих ИТК. В его пользу говорит недостаточно углубленное изучение оптимальных режимов дозирования для опухолей с мутацией EGFR. Математическое моделирование показывает, что различные схемы дозировки могут положительно влиять на развитие устойчивости (пролонгировать резистентность) без ущерба для эффективности [25].
При уже имеющейся вторичной резистентности к ИТК целесообразно назначение дополнительной терапии, ориентированной на подавление механизма лекарственной устойчивости.
Как минимум две возможности комбинирования терапии ИТК предложены на сегодняшний день, и в первую очередь, при помощи препаратов группы ингибиторов сигнальной трансдукции. Эта схема может применяться при лечении опухолей с подтвержденной мутацией EGFR наряду с избыточной экспрессией и/или аномальной активацией другого фактора роста или рецепторной системы (например, IGFR-I). В таких случаях комбинация ИТК с ингибиторами сигнальной трансдукции (например, PI3K) может обеспечить более длительный ответ на терапию. Практическая проблема состоит в том, что ингибиторы PI3K имеют целый ряд токсических побочных эффектов из-за существенной роли PI3K в физиологических процессах.
Вторая возможность преодолеть или пролонгировать развитие приобретенной резистентности к ИТК состоит в их комбинации с таргетными антиангиогенными препаратами. Этот способ представляется перспективным как минимум по двум причинам. В первую очередь, его применение сопряжено с феноменом «двойного удара», при котором выживаемость пациентов обусловлена элиминацией эндотелиальных клеток в дополнение к непосредственной антиопухолевой активности ИТК. К тому же снижение порога выживаемости опухолевых клеток вследствие ингибирования EGFR может способствовать повышению восприимчивости опухоли к антиангиогенной терапии [26]. Таким образом, комбинирование ИТК и антиангиогенных препаратов может повышать эффективность терапии, уменьшая вероятность выживания и экспансии резистентных популяций опухолевых клеток.
Успех комплексной терапии резистентности к ИТК зависит от возможности быстро определить механизм устойчивости. С этой целью пациенты с НМРЛ и мутацией EGFR в опухолевых клетках, у которых терапия ИТК была эффективной, при развитии лекарственной резистентности должны подвергнуться повторной биопсии или определению циркулирующей опухолевой ДНК в плазме с целью точного анализа механизма устойчивости.
Проведение повторных биопсий затруднено вследствие инвазивности процедуры, а также вероятности внутриопухолевой гетерогенности, которая может привести к получению некорректного результата [27]. Исследования показывают, что геномные изменения в солидных опухолях могут быть охарактеризованы с помощью массивного параллельного секвенирования циркулирующих внеопухолевых молекул ДНК, высвобожденных из опухолевых клеток. Этот сравнительно экономичный и быстрый метод, получивший название неинвазивной жидкостной биопсии, может значительно облегчить анализ механизма резистентности [28].
Неинвазивным методом – выделением опухолевой ДНК [29], а также циркулирующих опухолевых клеток из плазмы [30] – может быть успешно выявлена мутация Т790М. Неинвазивный анализ приобретенной резистентности путем выделения ДНК в плазме результативен и при других механизмах развития резистентности [31].
Молекулярно-генетическое тестирование в плазме, с помощью которого можно определить и качественное, и количественное содержание мутаций, сопряженных с резистентностью к ИТК, позволяет дать оценку эффективности терапии ИТК и течению заболевания. Так, в российском исследовании Д.Д. Сакаевой и М.Г. Гордиева [32], анализировавшем динамику мутаций в гене EGFR в цоДНК при распространенном немелкоклеточном раке легкого на фоне терапии гефитинибом, было показано, что объективный ответ на терапию коррелировал с исчезновением активирующих мутаций EGFR в плазме. Повторное же появление в плазме крови активирующих мутаций EGFR и/или Т790М являлось одним из предикторов прогрессии НМРЛ.
Осимертиниб: таргетная терапия Т790М-позитивного НМРЛ
Высокая частота активирующих мутаций, сопряженных с резистентностью к ИТК EGFR, обусловила активизацию поиска новых таргетных препаратов, которые помогли бы решить проблему устойчивости. Определенно самым значимым достижением в терапии Т790М-позитивного НМРЛ последних лет стало создание первого ИТК третьего поколения, действующего как на активирующую мутацию, так и на мутацию вторичной резистентности Т790М в гене EGFR, – осимертиниба. Осенью 2017 года препарат Тагриссо® (осимертиниб) был зарегистрирован в РФ для терапии местнораспространенного или метастатического НМРЛ с мутацией Т790М в гене EGFR.
Осимертиниб представляет собой необратимый ингибитор ТК EGFR, эффективный при наличии сенсибилизирующих мутаций гена EGFR и мутации Т790М, связанной с развитием резистентности к ИТК [33]. Препарат селективно действует на клетки как с активирующими мутациями гена EGFR (мутация L858R в 21-м экзоне, делеции в 19 экзоне), так и с мутацией резистентности Т790М, являющейся основной причиной резистентности к ИТК EGFR 1-2 поколений.
Осимертиниб отличается высокой активностью и ингибирующим действием в отношении EGFR во всех клинически значимых линиях НМРЛ, несущих сенсибилизирующие мутации EGFR и мутацию резистентности Т790М, что приводит к подавлению клеточного роста [33]. При этом препарат демонстрирует низкую активность в отношении клеточных линий, имеющих дикий тип EGFR (IC50 461-650 наномоль для различных клеточных линий).
Основанием для одобрения препарата стали результаты рандомизированного клинического исследования III фазы AURA3 [34] с участием 419 пациентов с Т790М-позитивным НМРЛ, прогрессирующим после терапии первой линии ИТК EGFR. В ходе исследования осимертиниб продемонстрировал статистически значимое преимущество по эффективности в сравнении с комбинацией пеметрексед/платина по показателю выживаемость без прогрессирования, составившему 10,1 месяца на осимертинибе в сравнении с 4,4 месяца на химиотерапии (ОР 0,3; 70% снижение риска; 95% ДИ: 0,23-0,41; p<0,001) [34].
Кроме того, пациенты, получавшие Тагриссо®, имели высокую частоту объективного ответа на терапию (71%), и у 9 из 10 из них удалось достичь контроля над заболеванием. При этом препарат продемонстрировал благоприятный профиль безопасности, что позволяет поддерживать хорошее качество жизни у довольно тяжелой категории пациентов с прогрессированием НМРЛ на фоне терапии ИТК EGFR 1-2 поколения.
По сути, появление нового эффективного таргетного препарата для пациентов с прогрессирующим НМРЛ означает изменения в алгоритмах диагностики и терапии заболевания. Пациентам с прогрессированием на фоне терапии ИТК EGFR показано проведение повторного молекулярно-генетического тестирования для определения молекулярного профиля опухоли и выбора дальнейшей тактики терапии. Так, в руководстве Европейского общества медицинской онкологии (ESMO) и Всеобщей национальной онкологической сети (NCCN) по диагностике, лечению и наблюдению больных раком легкого указано, что для пациентов с клиническим прогрессированием на фоне предыдущего лечения ингибиторами тирозинкиназ EGFR 1-2 поколений и подтвержденной мутацией Т790М должно быть предложено лечение осимертинибом в дозе 80 мг в сутки [35, 36].
Отечественные клинические рекомендации и руководства по лекарственному лечению злокачественных опухолей АОР рекомендуют проведение таргетной терапии осимертинибом до прогрессирования в качестве 2-й линии НМРЛ после прогрессирования на фоне приема ингибитора тирозинкиназы EGFR в случае выявления мутации Т790М в гене рецептора эпидермального фактора роста (EGFR) [37].
Вывод
Ингибиторы ТК, как препараты молекулярной таргетной терапии, открыли новые перспективы в лечении НМРЛ с активирующими мутациями EGFR. Однако генетическая нестабильность опухолевых клеток приводит к развитию приобретенной резистентности к ИТК. Преодоление устойчивости опухоли – серьезный вызов, который стоит перед современными учеными. Определенные положительные результаты в борьбе с лекарственной устойчивостью, которые продемонстрировали новые стратегии дозирования, а также комбинированная терапия, позволяют смотреть в будущее с некоторой долей оптимизма.
Список литературы
- Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2015. CA. Cancer J. Clin. 2015. 65, P. 5-29.
- Schiller J.H., et al. Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N. Engl. J. Med. 2002. 346, P. 92-98.
- Keedy V.L., et al. American Society of Clinical Oncology provisional clinical opinion: Epidermal growth factor receptor (EGFR) mutation testing for patients with advanced non-small-cell lung cancer considering first-line EGFR tyrosine kinase inhibitor therapy. J. Clin. Oncol. 2011. 29, P. 2121-2127.
- Lynch T.J., et al. Activating Mutations in the Epidermal Growth Factor Receptor Underlying Responsiveness of Non-Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2004. 350, P. 2129-2139.
- Kim E.S., et al. Gefitinib versus docetaxel in previously treated non-small-cell lung cancer (INTEREST): a randomised phase III trial. Lancet. 2008. 372, P. 1809-1818.
- Pao W., Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat. Rev. Cancer. 2010. 10, P. 760-774.
- Greulich H., et al. Oncogenic transformation by inhibitor-sensitive and-resistant EGFR mutants. PLoS Med. 2005. 2, e313.
- Kosaka T., et al. Analysis of epidermal growth factor receptor gene mutation in patients with non-small cell lung cancer and acquired resistance to gefitinib. Clin. Cancer Res. 2006. 12, P. 5764-9.
- Kobayashi S., et al. EGFR Mutation and Resistance of Non-Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2005. 352, P. 786-792.
- Costa D.B., Schumer S.T., Tenen D.G., Kobayashi S. Differential responses to erlotinib in epidermal growth factor receptor (EGFR)-mutated lung cancers with acquired resistance to gefitinib carrying the L747S or T790M secondary mutations. J. Clin. Oncol. 2008. 26, P. 1182-1184.
- Balak M.N., et al. Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin. cancer Res. 2006. 12, P. 6494-6501.
- Bean J., et al. Acquired resistance to epidermal growth factor receptor kinase inhibitors associated with a novel T854A mutation in a patient with EGFR-mutant lung adenocarcinoma. Clin. cancer Res. 2008. 14, P. 7519-7525.
- Engelman J.A., et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007. 316, P. 1039-1043.
- Cipriani N.A., Abidoye O.O., Vokes E., Salgia R. MET as a target for treatment of chest tumors. Lung Cancer. 2009. 63, P. 169-179.
- Bladt F., Riethmacher D., Isenmann S., Aguzzi A., Birchmeier C. Essential role for the c-met receptor in the migration of myogenic precursor cells into the limb bud. Nature. 1995. 376, P. 768-771.
- Takezawa K., et al. HER2 amplification: a potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov. 2012. 2, P. 922-933.
- Morinaga R., et al. Sequential occurrence of non-small cell and small cell lung cancer with the same EGFR mutation. Lung Cancer. 2007. 58, P. 411-413.
- Frederick B.A., et al. Epithelial to mesenchymal transition predicts gefitinib resistance in cell lines of head and neck squamous cell carcinoma and non-small cell lung carcinoma. Mol. Cancer Ther. 2007. 6, P. 1683-1691.
- Uramoto H., et al. Epithelial-mesenchymal transition in EGFR-TKI acquired resistant lung adenocarcinoma. Anticancer Res. 2010. 30, P. 2513-2517.
- Guix M., et al. Acquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGF-binding proteins. J. Clin. Invest. 2008. 118, P. 2609-2619.
- Yun C.-H., et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. 2008. 105, P. 2070-2075.
- Mulloy R., et al. Epidermal growth factor receptor mutants from human lung cancers exhibit enhanced catalytic activity and increased sensitivity to gefitinib. Cancer Res. 2007. 67, P. 2325-2330.
- Mok T.S. Living with imperfection. J. Clin. Oncol. 2010. 28, P. 191-192.
- Riely G.J., et al. Prospective assessment of discontinuation and reinitiation of erlotinib or gefitinib in patients with acquired resistance to erlotinib or gefitinib followed by the addition of everolimus. Clin. Cancer Res. 2007. 13, P. 5150-5.
- Foo J., Michor F. Evolution of resistance to targeted anti-cancer therapies during continuous and pulsed administration strategies. PLoS Comput Biol. 2009. 5, e1000557.
- Rak J., Joanne L.Y., Kerbel R.S., Coomber B.L. What do oncogenic mutations have to do with angiogenesis/vascular dependence of tumors? Cancer Res. 2002. 62, P. 1931-1934.
- Gerlinger M., et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. N. Engl. J. Med. 2012. 366, P. 883-892.
- Forshew T., et al. Noninvasive Identification and Monitoring of Cancer Mutations by Targeted Deep Sequencing of Plasma DNA. Sci. Transl. Med. 2012. 4, 136ra68.
- Kuang Y., et al. Noninvasive Detection of EGFR T790M in Gefitinib or Erlotinib Resistant Non-Small Cell Lung Cancer. Am. Assoc. Cancer Res. 2009. 15, P. 2630-2636.
- Maheswaran S., et al. Detection of Mutations in EGFR in Circulating Lung-Cancer Cells. N. Engl. J. Med. 2008. 359, P. 366-377.
- Murtaza M., et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. 2013. 497, P. 108-112.
- Сакаева Д.Д., Гордиев М.Г. Анализ динамики мутаций в гене EGFR в цоДНК при распространенном немелкоклеточном раке легкого на фоне терапии гефитинибом. Российский онкологический конгресс, 2015.
- Инструкция по применению лекарственного препарата Тагриссо.
- Mok T.S., et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N. Engl. J. Med. 2017. 376, P. 629-640.
- Kerr K.M., et al. Second ESMO consensus conference on lung cancer: pathology and molecular biomarkers for non-small-cell lung cancer. Ann. Oncol. 2014. 25, P. 1681-1690.
- Ettinger D., Wood D., Akerley W. NCCN guidelines insights: non-small cell lung cancer, version 4.2016. J. Natl. 2016.
- Клинические рекомендации. Рак легкого Портал Российского общества «Ассоциация онкологов России». 2017. (Accessed: 11 December 2017).

