
Современные подходы к лечению ретинобластомы
С.В. Саакян
ФГУ «МНИИ Глазных болезней им. Гельмгольца Росмедтехнологий»,
отдел офтальмоонкологии и радиологии, г. Москва
Опубликовано в «Российском офтальмологическом журнале», том 1, №1, 2008
Введение
Ретинобластома (РБ) – наиболее распространенная внутриглазная злокачественная опухоль оптической части сетчатки нейроэктодермального происхождения. Опухоль развивается внутриутробно или в раннем возрасте и характеризуется высокой степенью злокачественности, инвазивностью, способностью быстро метастазировать в соседние органы и ткани. В последние годы наблюдается рост частоты РБ в популяции: в настоящее время она составляет 1:10000-20000 живых новорожденных [2,7,16], хотя еще 20 лет назад частота опухоли оценивалась как 1:30000 [1,3]. Участие генетических факторов в процессе канцерогенеза в настоящее время не подвергается сомнению. Многочисленными исследованиями подтверждено, что злокачественная опухоль развивается в результате как структурных, так и функциональных (эпигенетичеких) мутаций генов.
Ретинобластома – наследственное заболевание, и предрасположенность к ней обусловлена герминальной мутацией в одном из аллелей гена RB1, который относится к классу генов-супрессоров опухолевого роста и расположен в проксимальном районе длинного плеча хромосомы 13 (q14.1). Передается потомству по аутосомно-доминантному типу с варьированием экспрессивности и достаточно высокой пенетрантностью (90%) [5,6]. РБ – первое онкологическое заболевание, на примере которого была сформулирована двухударная теория канцерогенеза, предполагающая, что для перехода нормальной клетки в опухолевую необходимы два последовательных мутационных события [5,10]. При наследственной форме РБ вторая мутация может появиться в любой соматической клетке организма, поэтому у больных с герминальной мутацией в гене RB1 существует большой риск развития различных злокачественных опухолей, таких как остеосаркома, рак молочной железы и другие. Существует спорадическая и наследственная форма заболевания. Спорадическая форма встречается в 60% случаев и проявляется как монолатеральная, монофокальная опухоль у детей после года. Наследственная форма встречается в 40% случаев, выявляется преимущественно на первом году жизни и сопровождается двусторонним поражением с мультицентричным характером роста.
РБ развивается в возрасте от 0 до 9 лет. У подавляющего большинства детей РБ развивается в возрасте до 3 лет. Однако в последние 10 лет наблюдается увеличение заболеваемости у детей старше 5 лет. Выявление опухоли в юношеском возрасте считается раритетом [6,10]. Оба глаза поражаются одинаково часто. Различий по половому признаку, расовой принадлежности не установлено. В начале 20 века дети, страдающие от ретинобластомы, имели мало шансов на выживание. Прогресс в лечении этого заболевания позволил повысить выживаемость с 30% в 1930 году, до 80% в 1960 г. В настоящее время, благодаря внедрению в практику различных комбинированных методов лечения, эта цифра достигает 95% в развитых странах [10,16]. Ключевую роль в революционном снижении смертности сыграли усовершенствование лучевых технологий и внедрение новых лекарственных препаратов, которые были включены в протоколы лечения детей [3].
Целью настоящего исследования является анализ эффективности проводимого комбинированного лечения больных с различными формами ретинобластомы.
Материал и методы
Проведен ретроспективный анализ эффективности лечения детей с различными формами РБ, поступившими в стационар отдела офтальмоонкологии и радиологии МНИИ ГБ им. Гельмгольца с января 2002 г. по декабрь 2006 г. Под наблюдением находились 164 ребенка (79 девочек и 85 мальчиков) с первичной ретинобластомой. Монокулярная форма выявлена у 110 больных (67.0%), бинокулярная – у 54 детей (32.9%), у 1 ребенка выявлена трилатеральная ретинобластома. Наследственная РБ отмечена в 9 случаях (5.5%). Средний возраст больных с монокулярной формой заболевания при обращении составил 28±2.4 мес., при бинокулярном поражении – 14.5±1.5 мес. До госпитализации дети проходили полноценное общеклиническое обследование на предмет исключения регионарных и отдаленных метастазов. Всем детям после года проводили КТ орбит и головного мозга, дети до года подвергались этому обследованию в случае подозрения на экстрабульбарный рост или наличие метастазов в головной мозг. В стационаре все больные были обследованы в условиях медикаментозного сна с максимальным мидриазом, во время которого проводилось полное офтальмологическое обследование, включающее в себя, помимо рутинных методов, осмотр глазного дна ретинальной педиатрической камерой (RetCam), УЗИ, УЗДГ и ОСТ. Согласно классификации ВОЗ, III стадия заболевания была выявлена у 80% детей, IV стадия – в 11% случаев, а I-II стадия присутствовала только у 9% больных. Согласно принятой в западных странах классификации Reese-Ellsworth, построенной по прогностическому принципу, 75% больных отнесены к 4-5 группе (плохой прогноз), а 25% – к 1-3 группе.
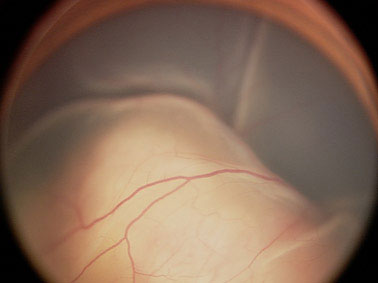
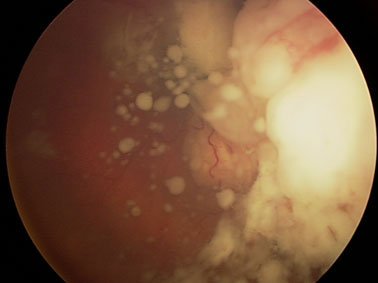
В клинической картине монокулярных РБ преобладали больные со смешанным (рис.1) и эндофитным характером роста (74%), а при бинокулярном поражении – в лучших глазах наблюдался экзофитный (рис.2) или эндофитный (рис.3) характер роста (93%). У 36 из 54 больных (66.7%) с бинокулярной РБ имело место мультифокальное поражение сетчатки (рис.4). Количество опухолевых очагов колебалось от 2 до 5. У большинства больных с монокулярным поражением (82%) параметры опухоли определить было невозможно, так как опухоль выполняла практически весь глаз, помимо этого, отслойка сетчатки, кровоизлияния препятствовали детальному осмотру опухоли. Кальцификаты выявлялись в 76% случаев. При бинокулярном поражении толщина опухоли в «лучших» глазах варьировала от 1.2 до 8.5 мм (в среднем 5.3±0.7мм), а диаметр основания – от 3.5 до 14.5 мм (в среднем 7.5±1.2 мм).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Всем больным проведено комбинированное лечение с обязательной полихимиотерапией. Ликвидационные операции при монокулярном поражении сопровождались адъювантной полихимиотерапией по дробно-протяженной схеме, которая проводилась 1 раз в 3 недели в течение года под контролем гематологов и педиатров. Протокол включал внутривенное введение винкристина в дозе 0.05 мг/кг веса и циклофосфана из расчета 30 мг/кг веса. При проведении органосохраняющих операций лечение начинали с неадъювантной полихимиотерапии, предложенной нами, включающей цитостатик платинового ряда – карбоплатин (доза 18.7 мг/кг веса) и винкристин (0.05 мг/кг веса) в/в 1 раз в 3 недели с соблюдением всех норм введения. Количество курсов колебалось от 3 до 11 (стандартный блок – 6 курсов), зависело от параметров опухоли и ее чувствительности. После уменьшения параметров опухоли переходили к одному из видов локального разрушения опухоли (лазеркоагуляция, криодеструкция, брахитерапия Ru106+Rh106 или Sr90+Y90). Сроки наблюдения за больными составили от 12 до 60 мес., в среднем 42.5 мес. При оценке эффективности проведенной терапии из исследования были исключены 8 детей с бинокулярной РБ, наблюдение за которыми по не зависящим от нас причинам было прервано на разных этапах лечения.
Результаты и обсуждение
Анализ полученных результатов показал, что единственным методом лечения, который можно было предложить 91% больных (100 детей) с монокулярной ретинобластомой, являлась энуклеация. Во всех случаях опухоль или выполняла практически всю полость глаза, или имелись вторичные осложнения в виде тотально отслоенной сетчатки, вторичной глаукомы, гемофтальма, диссеминации опухолевых клеток в стекловидное тело или инвазия в ДЗН (рис.5), распространение опухоли в переднюю камеру (рис.6). Объясняется это, во-первых, поздним обращением больных к врачу, а во-вторых, отсутствием системы активного выявления врожденной патологии у детей младшего возраста. Известно, что самой распространенной жалобой является свечение зрачка (симптом «кошачьего глаза», лейкокория – 68%) однако этот признак свидетельствует о поздней стадии заболевания, при которой сохранение глаза практически невозможно. Анализ предъявляемых родителями жалоб свидетельствует о том, что второй основной жалобой у детей младшего возраста является косоглазие (32%). Но, к сожалению, врачи, как правило, не обращают на него внимания, в то время как косоглазие может возникать на ранней стадии развития центрально расположенной опухоли. Об отсутствии настороженности у офтальмопедиатров в плане выявления РБ говорит тот факт, что даже в семьях с высоким риском появления больного ребенка к врачу с просьбой об обследовании обращались родители [6].
{kind=link}
{kind=link}
В некоторых работах дискутируется вопрос о необходимости проведения адъювантной ПХТ после ликвидационных операций [7]. Основанием для исключения ПХТ из протокола лечения является отсутствие прорастания опухоли в хориоидею. Однако согласно полученным в нашем исследовании патоморфологическим результатам у 89% больных с рТ3с стадией развития опухоли наблюдались признаки инвазии опухоли в подлежащую хориоидею, что, как известно, резко повышает риск метастазирования опухоли и считается плохим прогностическим признаком [9]. Следовательно, включение в протокол лечения адъювантной полихимиотерапи, направленной на эрадикацию микрометастазов и профилактику рецидивов заболевания, является вполне обоснованным и необходимым принципом. В 10 случаях (9.09%), несмотря на большие размеры опухоли (более 8 мм), была предпринята попытка органосохраняющего лечения, которое начинали с неоадъювантной ПХТ. Глаза удалось сохранить у 8 больных, что составило 7.2% от общего числа больных с монокулярной формой опухоли.
Основной группой больных, среди которых развернулась борьба за сохранение глаза и зрения, были дети с бинокулярным поражением. После исключения 8 детей анализ эффективности лечения был проведен среди 46 детей с двухсторонней ретинобластомой. Необходимо отметить, что после тщательного обследования у 3 детей было выявлено полное поражение обоих глаз с развитием вторичной глаукомы, в связи с чем произведена одномоментная бинокулярная энуклеация. Еще у четырех детей была предпринята попытка сохранения второго глаза, однако, несмотря на временный эффект после неоадъювантной ПХТ, брахитерапии и лазеркоагуляции, возникли рецидивы опухоли и новые очаги, которые вынудили нас провести энуклеацию и второго глаза. Таким образом, на фоне лечения было удалены оба глаза в 8.7% случаев. У двух детей, благодаря предпринятым мерам, удалось сохранить оба глаза (4.3%). У 39 больных после обследования худшие глаза были удалены (Т3сm). Парный «лучший» глаз был подвергнут комбинированному лечению, на первом этапе которого применялась НХТ, разрушающая первичную опухоль до размеров, позволяющих проводить один из методов локального разрушения. Во всех случаях вторым этапом проводилась брахитерапия. При этом у 8 детей (20.5%) на фоне лечения возникли рецидивы или новые очаги, по поводу которых производилась повторная брахитерапия или разрушающая лазеркоагуляция. Помимо локального облучения, в случаях с мультицентричным поражением, при небольших размерах (толщиной до 1.5 мм), очаги, расположенные постэкваториально, разрушались с помощью диодного лазера (18 детей – 46.2%), а преэкваториальные – с помощью криодеструкции (3 больных). Таким образом, благодаря многофакторному воздействию на мультицентричные опухолевые очаги удалось сохранить 39 из 46 глаз (84.8%), а предметное зрение – у 35 детей (рис.7).
{kind=link}
Анализ результатов комбинированного лечения больных с РБ еще раз доказал, что разрушение опухоли с помощью локального облучения офтальмоаппликаторами является наиболее эффективным методом. Известно, что брахитерапия для лечения ретинобластомы впервые была использована Stallard and Moore в 1920 г. в виде капсул с радоном. Затем радиоактивные источники неоднократно пересматривались. Сейчас в мире используются в основном офтальмоаппликаторы с радиоактивным йодом (125I) и рутением 106Ru. Локальное облучение рутениевыми аппликаторами впервые было применено в 1960 г. немецкими офтальмологами Lommatzsch and Vollmar. У нас в стране брахитерапия при РБ впервые использована Г.Д. Зарубеем в 1978 г. в отделении патологии глаз у детей МНИИ ГБ им. Гельмгольца (руководитель – проф. А.В. Хватова). Несмотря на то, что радиоактивный йод обладает большей проникающей способностью, чем рутений, применение его ограничено быстрым распадом частиц (одноразовое применение), большим количеством осложнений и дороговизной. Рутениевые офтальмоаппликаторы зарекомендовали себя как надежные, хорошо переносимые при правильном расчете дозы источники. В нашем исследовании не было ни одного случая склеромаляции, несмотря на то, что в 8 случаях аппликатор был использован повторно.
Хорошие результаты, полученные при лечении детей с тяжелой формой поражения, объясняются включением в комплекс лечения обязательной полихимиотерапии. Протокол адъювантной полихимиотерапии, применяемой в отделе офтальмоонкологии и радиологии МНИИ ГБ им. Гельмгольца, разработан в 1989 г. Международным комитетом по изучению ретинобластомы во главе с J. Shields. Почти 20-летний опыт использования подтвердил его высокую эффективность и хорошую переносимость. Внедрение в практику в 1995 г. неоадъювантной терапии произвело революцию в лечении больших и мультицентричных опухолей, так как основной компонент ПХТ – карбоплатин – обладает способностью разрушать первичную опухоль внутри глаза. Необходимо отметить, что приведенный нами протокол можно и нужно использовать только при локальных формах опухоли. При наличии признаков экстрабульбарного роста или генерализации процесса дети направляются в общеонкологические клиники для проведения высокодозной химиотерапии [3,7,13].
Благодаря карбоплатину стало возможным сохранять глаза, которые ранее однозначно подлежали удалению или подвергались наружному облучению в виде дистанционной гамматерапии. Безусловно, рентгенотерапия позволила в середине прошлого века резко повысить непосредственные результаты лечения детей с ретинобластомой и уменьшить смертность от заболевания. Однако уже в 1969 г. Sagerman [14] показал высокую зависимость развития вторичных опухолей от облучения. В последующие годы многие авторы подтверждали эту взаимосвязь. Так, Abramson [8] описал многочисленные случаи развития вторых, третьих и четвертых опухолей у выживших после РБ больных, причем количество вторых опухолей увеличивалось с каждым прожитым десятилетием, а смертность возрастала. Основываясь на этих данных, мы резко сократили показания к облучению детей. К сожалению, полностью исключить дистанционную гамма-терапию из протокола лечения невозможно, так как вероятность рецидива опухоли в орбиту или продолженного роста в полость мозга при экстрабульбарном росте очень велика. В связи с этим при наличии клеток в ретробульбарном пространстве или на срезе зрительного нерва больные направлялись на наружное облучение. В нашей серии мы не наблюдали развития вторых или вторичных опухолей, но косметические дефекты развились у всех 7 детей, направленных на облучение.
За исследуемый период (январь 2002 – декабрь 2006 гг., 5 лет) в общей группе погибло 4 ребенка, что составило 2.6%. Во всех случаях дети погибли от прорастания опухоли в головной мозг. Смертность в группе с монокулярным поражением составила 1.8%, а смертность в группе с бинокулярным поражением – 4.3%.
Таким образом, благодаря проведению адекватного индивидуального комбинированного и многофакторного лечения различных форм ретинобластомы выживаемость детей с бинокулярной ретинобластомой составила 95.7%, а вторые глаза и зрение сохранены в 84.7% случаев.
Все выжившие дети, несмотря на то, что являются инвалидами с детства, благодаря даже частично сохраненному зрению полностью адаптированы в окружающую общественно-социальную среду. Они посещают детские и средние образовательные учреждения, что свидетельствует о большой медико-социальной значимости разработанного комплекса комбинированного лечения больных с разными формами РБ и повышения качества жизни не только самих детей, но и их родителей.
Выводы
Выявляемость ретинобластомы на ранних стадиях развития остается чрезвычайно низкой (4%). Своевременная диагностика ретинобластомы позволяет сохранить не только глаз, но и зрение. С целью выявления опухоли на ранних стадиях следует проводить офтальмоскопию с широким зрачком всем детям при рождении, в 3, 6 и 12 мес. Далее не реже, чем раз в год. Дети из группы риска должны находиться под постоянным динамическим наблюдением и обследоваться каждые 3 мес. Внедрение в клинику протокола неоадъювантной полихимиотерапии и многофакторного воздействия на опухоль позволило расширить показания к органосохранному лечению больших и мультицентричных ретинобластом, повысило выживаемость и качество жизни детей и их семей.
Литература
- Аветисов Э.С., Ковалевский Е.И., Хватова А.В. Руководство по детской офтальмологии.// М.: Медицина, 1987, С.440.
- Бровкина А.Ф. Офтальмоонкология: Руководство для врачей.// М.: Медицина, 2002, С.315-328
- Дурнов Л.А. Руководство по детской онкологии.// М.: Миклош, 2003, С.504.
- Дыбов Ст. Ретинобластома.// София, 1975, С.126.
- Залетаев Д.В. Современная молекулярная генетика в офтальмологии. Наследственные и врожденные заболевания сетчатки и зрительного нерва.// Под ред. А.М. Шамшиновой. Медицина, 2001, С.14-23.
- Саакян С.В. Ретинобластома (клиника, диагностика, лечение).// М.: ОАО «Изд-во Медицина», 2005, С.200.
- Ушакова Т.Л., Максимова О.В., Долгополов И.С., Глеков И.В. и др. Результаты лечения ретинобластомы высокого риска.// Сб. трудов науч.-практ. конф. «Современные технологии в диф. диагностике и лечении внутриглазных опухолей», М., 2007, С.270-273.
- Abramson DH, Frank CM Second nonocular tumors in survivors of bilateral retinoblastoma: a possible age effect on radiation-related risk.// Ophthalmology, 1998, vol.105, №4, pp.573-57.
- Acquaviva A, Ciccolallo L, Rondelli R et al. Mortality from second tumour among long-term survivors of retinoblastoma: a retrospective analysis of Italian retinoblastoma registry.// Oncogene, 2006, 25, pp.5350-5357.
- Balmer A, Zografos L, Munier F. Diagnosis and current management of retinoblastoma.// Oncogene, 2006, 25, pp.5341-5349.
- Lommaizsch P, Vollmar R. A new way in the conservative therapy of intraocular tumors by means of β-irradiation (ruthenium 106) with preservation of vision.// Klin Monatsbl Augenheilkd, 1966, 148:682-699.
- Moore RF, Stallard HB, Milner JG. Retinal glioma treated by radon seeds.// Br J Ophthalmol, 1931; 15:673-696.
- Namouni F, Doz F, Tanguy ML et al. High-dose chemotherapy with Carboplatin, Etoposide and Cyclophosphamide followed by a haematopoietic stem cell rescue in patients with high-risk retinoblastoma: a SFOP and SFGM study.// Europ. J. of Сancer, 1997, vol.33, №14, pp.2368-2375.
- Sagerman RH, Cassidy JR, Tretter P, Ellsworth RM. //Am J Roentgend Ther Nucl Med 1969, 105:529-535.
- Shields CL, Shields JA, Needle M et al Combined chemoreduction and adjuvant treatment for intraocular retinoblastoma.// Opthalmology, 1997, vol.104, №12, pp.2101-2111.
- Schueler AO, Fluhs D, Bornfeld N et al. β-Ray Brachytherapy with 106RU plaques for retinoblastoma.// Int. J.Radiation Оncology Biol. Phys., 2006, vol.65, №4, pp.1213-1221.